


近期,生物信息领域大神、美国丹娜-法伯癌症研究所李恒老师在Nature Reviews Genetics上发表了关于T2T基因组的综述文章《Genome assembly in the telomere-to-telomere era》,该文回顾目前在大型真核生物基因组高质量组装方面的实践,最终指向端粒到端粒(T2T)的组装。文章从6个方面对基因组T2T组装进行了总结,介绍常见T2T组装的数据类型,剖析最新的组装算法,解释评估组装的方法,讨论组装T2T基因组的挑战,为组装T2T基因组提供了自己的见解和指导。 本文先介绍前3个方面,以饕读者。
影响组装的基因组特性
决定基因组组装难易程度的主要因素不是基因组的大小,而是基因组的重复结构。重复序列可以通过比重复序列更长的reads来解析。不过,基因组一些特殊结构的重复区域比目前测序技术产生的reads要长得多,从而增加了组装的难度。然而,随着长片段重复序列区域的突变不断累积,很少有超过10kb的相同重复序列,再凭借高准确性的PacBio HiFi reads和ONT超长reads,就可以区分不同的重复序列,并成功地将它们组装起来。
重复序列可以大致分为三类:散在重复、串联重复和区段重复。
散在重复:主要是分散在基因组中的转座子,它们几乎都比现代长读长reads短,因此不再是组装的主要障碍。
串联重复:大多数染色体上的串联重复都比长读长reads短,因此也容易组装。然而,卫星重复是一种富集在着丝点上特别难以组装的超长串联重复,因为长读长reads无法跨越整个卫星阵列。
区段重复:在基因组中重复的非常长的DNA片段,通常比长读长和超长读长reads还要长,它们许多是串联成簇。如核糖体DNA(rDNA)可能包含高度相似拷贝的长串联阵列,长的rDNA阵列是最难组装的区域之一。
二倍体样本中的两个同源单倍型也可看作是彼此的重复,对于二倍体或多倍体样本来说,T2T的组装也意味着所有染色体都正确分相。能够解决相似重复序列的组装软件自然就有很强的能力分离同源单倍型。反之,无法进行单倍型分相的组装软件则无法解决相似重复拷贝的问题。虽然传统的组装算法会破坏同源单倍型,但目前的做法往往能在数百万个数据库中保留单倍型分相,并能在多种数据类型的情况下进行染色体规模的单倍型分相组装。
长读长和长距离测序技术
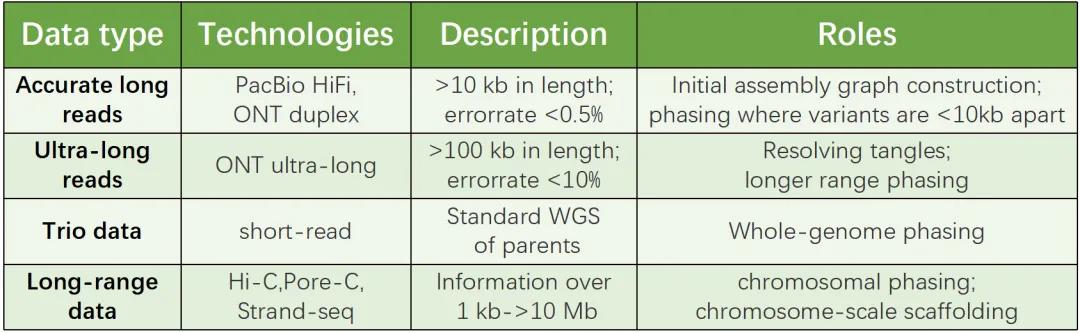
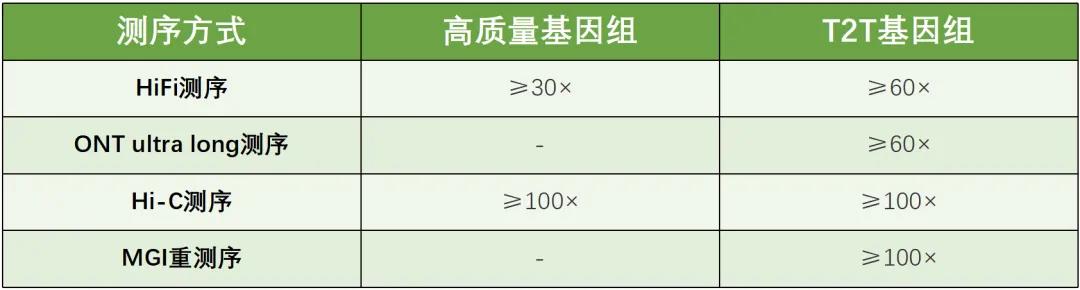
获得近T2T组装通常需要多种测序技术结合(表1)。长度长测序技术产生的序列长度通常≥10kb,当前主要以太平洋生物科学公司(PacBio)和牛津纳米孔技术公司(ONT)两家公司为代表。2019 年,PacBio推出了长度为10-20 kb且错误率低于0.5%的HiFi reads,是高质量组装的核心数据类型。市面上的ONT产品的准确率大致在90-95%,虽然错误率较高,但有一种ONT数据类型(超长reads)的长度≥100kb,有助于解决HiFi读数无法组装的重复序列的问题。ONT正在积极开发双链测序,准确度接近PacBio HiFi,长度也更长。一旦技术成熟,它将成为一种引人注目的数据类型。
即使是超长reads,其跨度也很少超过数百kb,为获得染色体长度的scaffold以及分相,需要长距离数据。使用最广泛的长距离数据类型Hi-C,它由两端可能来自同一染色体上的远距离的双端短reads(二代测序)组成,可以实现染色体分相和Mb距离contig的排序。Pore-C与Hi-C相似,但是用ONT测序。Strand-seq是另一种特别擅长染色体分相和contig排序的技术,但它更昂贵且很难在市场上获得。亲本测序数据或三联体(trio)数据具有强大的全基因组定相能力,也可以被视为一种长距离数据。
表1 长读长和长距离测序技术
近T2T组装方法
· 组装纯合基因组
对于纯合基因组,最可靠的T2T组装方案是同时使用PacBio HiFi和ONT超长测序(图1),首先使用HiFi reads构建初始组装图,该图由不包含确切的长重复序列的线性片段(unitigs)组成,它们之间可能存在连接,具体取决于重复结构。高度重复的区域表现为复杂的子图,称为 "tangles"。然后,将ONT超长reads锚定到unitigs上,并穿过"tangles",从而解决大部分tangles。超长reads还能修补偶尔出现的HiFi未覆盖率的组装间隙。当染色体在组装子图中分离不佳或不连续时,Hi-C数据将有助于生成染色体长度的scaffold。
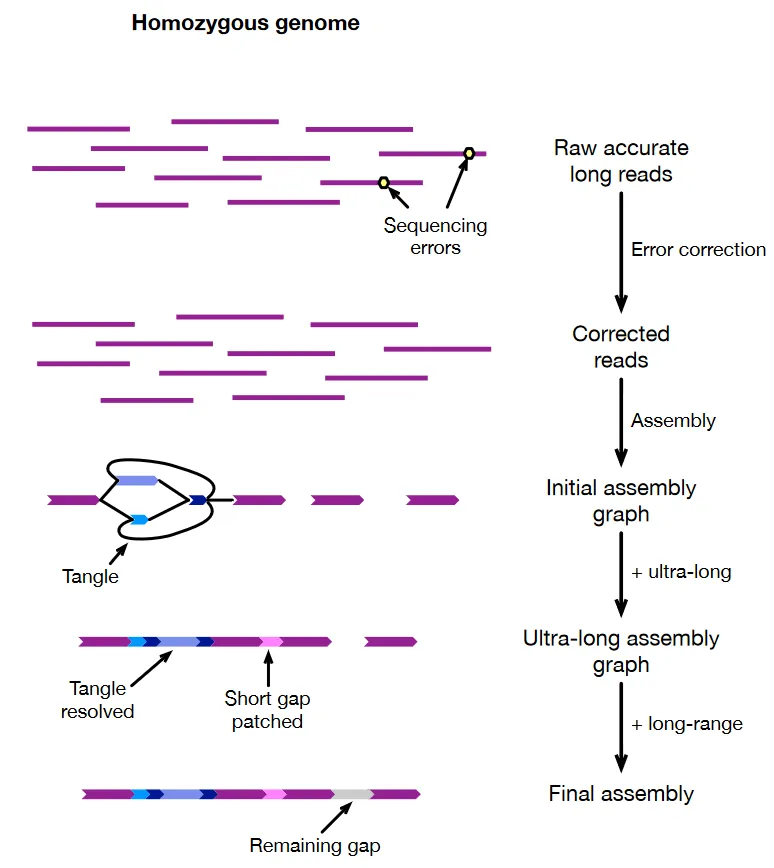
图1 纯合二倍体基因组的常见组装流程
目前,verkko和hifiasm可以整合PacBio HiFi和ONT超长数据,它们大致遵循图1中的工作流程,但在每个步骤中使用不同的算法。有时,仅使用HiFi数据即也可能实现纯合基因组的良好组装。Verkko、hifiasm、HiCanu和 LJA都可以单独使用HiFi reads实现多条人类染色体T2T组装。当需要搭建scaffold时,YaHS已取代SALSA成为Hi-C挂载的推荐方法,它是脊椎动物基因组计划(VGP)和达尔文生命之树计划(DToL)的首选scaffold搭建方法。
· 组装杂合二倍体基因组
组装杂合二倍体基因组的策略与纯合基因组类似(图2)。对于具有较长同源序列的基因组,包括人类基因组,仅使用HiFi和ONT超长组合可能无法对整个染色体进行分相。在这种情况下,建议使用能对整个基因组进行精确分相的三联体数据。当无法获得亲本样本时,可以使用Hi-C代替。Hi-C只能提供contig之间的相对分相信息,其功能不如三联体数据强大,尤其是在纠结的子图中。尽管如此,Hi-C仍然是可靠挂载染色体的关键数据类型。
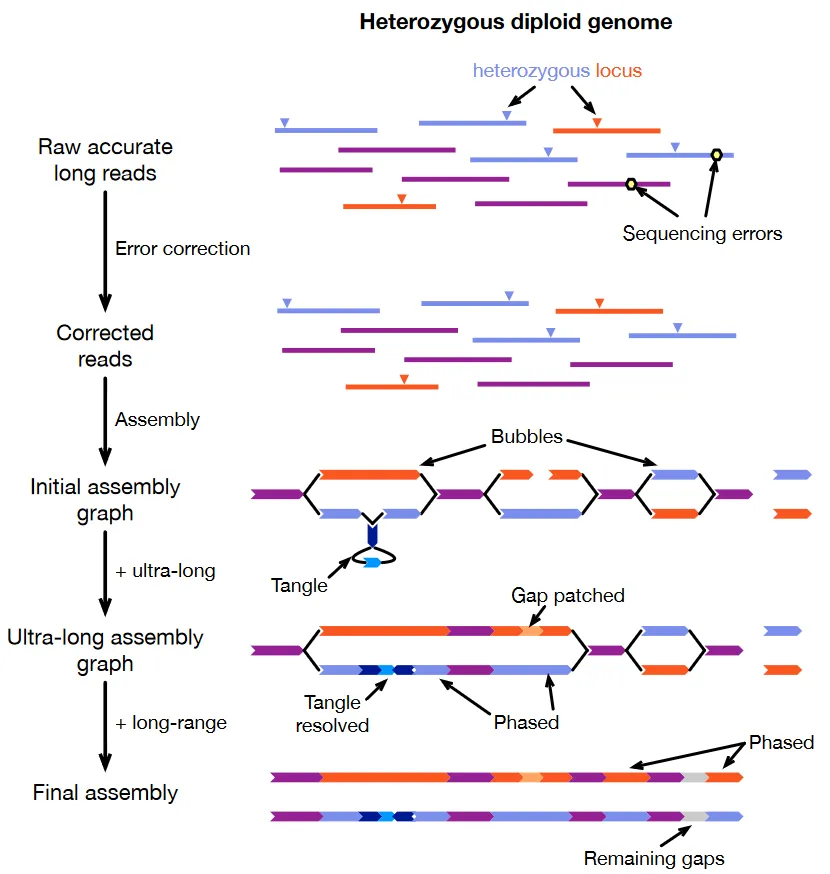
图2 杂合二倍体基因组的常见组装流程
就目前而言,ONT超长数据的获取成本相对较高,而且对DNA的需要量较大(通常为数十微克)。许多测序项目不会生成超长数据,仅利用HiFi数据就能生成primary/alternate组装对或双组装对(图3b、c)。为获得一个参考基因组,可能首选组装的primary序列,因为primary序列通常较长。alternate序列比较零碎,容易出错,在下游分析中通常会被忽略。双组装对代表了二倍体样本中的两个基因组,支持基于组装的变异检测,也支持使用双组装对构建泛基因组。不过,对于较短的contig,搭建scaffold可能会更加复杂。无论采用哪种方法,在区分旁系串联重复和同源单倍型重复时都会出现问题,尤其是在contig的末端,这可能会导致错误的重复。对于primary组装方法来说,这些问题中的许多(但不是全部)都可以通过启发式方法发现并解决,例如在purge_dups中实现的方法。
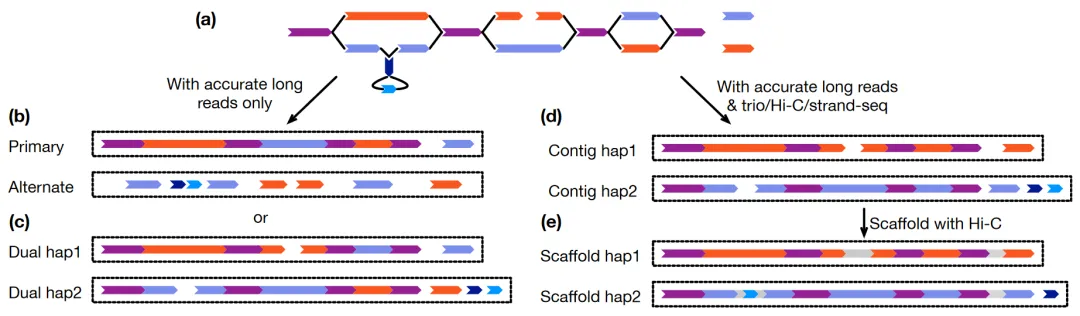
图3 二倍体分相组装的不同方法
将HiFi与trio、Hi-C或Strand-seq等长距离数据相结合,可以生成一对单倍型分辨率的组装对(图3d、e),这种组装对的连续性与双组装对相当。此外,它还保留了相位,并可通过Hi-C进一步将其挂载为分相的染色体。有研究表明,即使没有亲本数据,只要已知并足够频繁的标记去标记contig同源对,就可以利用印记甲基化标记确定这些染色体的亲本来源。
对于杂合基因组,verkko和hifiasm都能整合PacBio HiFi、ONT超长和长距离数据,并能组装多条单倍型分辨的人类T2T染色体。它们也可单独用于HiFi数据,并生成双组装对或primary组装。HiCanu也能利用HiFi数据生成primary组装,并达到相当的质量。
参考文献
Li, H., Durbin, R. Genome assembly in the telomere-to-telomere era. Nat Rev Genet (2024). https://doi.org/10.1038/s41576-024-00718-w

康普森农业长期致力于动植物基因组、泛基因组和T2T基因组组装研究,拥有丰富的项目方案设计及分析经验,涵盖家禽、家畜、粮食作物、园艺作物、花卉林木、水产等多样性物种,将为科研工作者提供全面的三代测序、基因组组装和群体重测序等方面的专业技术服务。
康普森基因组组装方案

天津:18710280840/022-24986099
北京:400 1869 509
邮箱:marketing@kangpusen.com
地址:北京市昌平区中关村生命科学园生命园路4号院4号楼7层
图文来源:北京康普森农业科技有限公司


 下载app
下载app